

Introduction
Materials and Methods
Pollen Collection
Pollen Germination
Colorimetric Assay using Congo Red
Flow Cytometry
Results and Discussion
Introduction
In vitro pollen germination on solidified media is still widely used to evaluate pollen viability. However, this method not only requires a lot of time and effort, it also has the disadvantage of poor accuracy and reproducibility (Cook and Stanley, 1960; Hartman et al., 2014). If the pollen grain is not uniformly distributed across the medium, then there is a tendency that the pollen germination rate of the low density portion is measured to be low (Brewbaker and Majumder, 1961).
To overcome these drawbacks, colorimetric methods for assessing pollen quality using tetrazolium salts (dehydrogenase), aniline blue (callose in pollen wall), acetocarmine (nucleic acid), Alexander’s stain (cytoplasmic constituents), and fluorescein diacetate (esterase) have been introduced (Oberle and Watson, 1953; Currier, 1957; Heslop-Harrison et al., 1984; Alexander, 1987; Heslop-Harrison, 1992). Colorimetric assays are often preferred, as these are faster and easier than in vitro pollen germination. However, they might obtain different results in some fruit species or cultivars (Oberle and Watson, 1953). Furthermore, they tend to overestimate viability, due to the presence of enzymes or other substances from non-viable pollen (Rodriguez-Riano and Dafni, 2000).
Congo Red (CR), a polysaccharide binding dye, has been used for histochemical observations of plant cell walls (Wood and Fulcher, 1978; Earp et al., 1983), and may interact with the cellulose surface through electrostatic forces, hydrogen bonding, or hydrophobic interactions (Yamaki et al., 2005). Recently, a semi-quantitative assay using CR staining for determining pollen tube extension was introduced (Hartman et al., 2014). This method has the advantage of performing “high-throughput screening” of certain molecules that modulate biological activity – that is, activity that either enhances or inhibits pollen germination – using a microtiter plate reader within a few hours. Although increases in CR absorbance indicate increases in pollen tube growth, this method may not be suitable for comparing the rate of germination among pollen with different viabilities.
The advantage of flow cytometry is that allows individual particles to be selectively discriminated based on the variable (i.e., cell size, internal complexity) being measured (Kerker, 1983; Givan, 2001). Both forward scatter (FSC) and side scatter (SSC) signals may provide information on the morphological differences among the cells, allowing cells within a heterogeneous population to be discriminated (Brown and Wittwer, 2000; Adan et al., 2017). Thousands of cells can be measured per second, with high accuracy and reproducibility (Bendall et al., 2012).
To verify pollen quality, it is essential to develop a reliable method that is able to complement the drawbacks of the conventional colorimetric method. Furthermore, the new method should be faster and more accurate than in vitro pollen germination. In this study, we show the advantages of colorimetric and cytometric analyses over the commonly used in vitro germination methods. The purpose of the present study is to establish a quantitative method for measuring pollen tube growth based on differences in CR absorbance or pollen distribution. To achieve this goal, 1) we compared the difference in CR absorbance between a positive control (PC), with pollen that does germinate, and negative control (NC), with pollen that does not germinate, after culturing the pollen. 2) The differences in pollen distribution between the PC and NC groups along the FSC and SSC parameters were investigated. 3) Whether the difference in either CR absorbance or pollen distribution between the PC and NC could be expressed as an equation was also examined.
Materials and Methods
Pollen Collection
Unopened flowers were collected in the balloon stage (5 days before full blooming) in April 2018 from pear trees of the “Wonwhang” cultivar growing in commercial orchards (Gongsan, Naju, Korea). The anthers of pear flowers that were not fully open were harvested and laid out on black kentpaper in a dehiscence room at a temperature of 20°C and relative humidity of 50% to dry and release their pollen over a period of 24 to 36 h. When more than 80% of the anthers were dehiscent, anthers were removed from the kentpaper and placed in a stainless steel bowl that contained a 100-mesh sieve (0.149 mm aperture). Acetone was poured into the stainless steel bowl, and the pollen was sieved gently through the 100-mesh sieve into an acetone solvent. Acetone supernatant was then discarded carefully from the stainless steel bowl, and the residual solvent was volatilized. Pollen was weighed and 10 g each were added to tightly sealed containers and deep frozen at -60°C until further use.
Pollen Germination
Unless otherwise stated, each assay was conducted by removing a pollen container from the deep freezer and allowing the pollen to thaw overnight at 4°C in a desiccator. Pollen was scattered uniformly onto a solidified medium containing 10% sucrose (w/v), 0.4 mM boric acid, 1 mM calcium nitrate, and 1% agar (Speranza et al., 2012), and then incubated at 25°C for 3 h. Pollen germination was recorded by capturing microscopic images every hour after the pollen finished incubating. A video camera (Axiocam ICc1, Carl Zeiss, Germany) connected to Axiostar Plus (Carl Zeiss, Germany) was used to acquire digital images of randomly selected fields in Petri dishes. A pollen grain was considered to have germinated when the length of its pollen tube was equal to or greater than the diameter of the pollen grain. Pollen germination was determined by counting three times from the digital images of at least 100 pollen grains for each group using ImageJ software (http://rsb.info.nih.gov/ij). The germination percentage was calculated as the proportion of pollen grains germinated to the total number of pollen grains observed.
The germination rate in liquid medium was measured by dividing pollen into positive control (PC) and negative control (NC) groups. The medium for the PC group for pollen germination contained 2% DMSO (v/v), and that for the negative control, which did not germinate, contained 10% DMSO (v/v) (Hartman et al., 2014). 2.5 mg·mL-1 of pollen was suspended in liquid medium containing 10% sucrose (w/v), 0.4 mM boric acid, 1 mM calcium nitrate, and 2% DMSO (for PC), or 10% DMSO (for NC), and then incubated at 25°C for 4 h. Pollen germination was recorded by photographing the pollen tubes every hour, without removing the germinated pollen from their growth dish, in a liquid medium 2 mm deep. The depth of the medium was slightly higher than that used by Read et al. (1993) for germinating tobacco pollen. Read et al. (1993) observed and immediately photographed the pollen tubes by culturing the pollen in a liquid medium 1.3-1.7 mm deep. Germinated pollen was counted germination percentage calculated using the same method as in solidified medium.
Colorimetric Assay using Congo Red
Germination and colorimetric assays of pear pollen divided into PC and NC groups were performed according to the method described by Hartman et al. (2014), with some modifications. Pollen suspension at a concentration of 1.25 to 10 mg·mL-1 was prepared in 1X pollen growth medium (PGM) containing 10% sucrose (w/v), 0.4 mM boric acid, and 1 mM calcium nitrate. To identify the difference in Congo Red (CR) absorbance between germinated and non-germinated pollen, DMSO at 2 or 10% (v/v) was added to the medium. 50 and 250 µL of DMSO was added to the test tubes of the PC and NC, respectively. Each tube was filled to a total volume of 625 µL by adding distilled water (575 µL for PC, 375 µL for NC). After adding 1,250 µL of pollen suspensions to each tube, 625 µL of 2X PGM – consisting of 20% sucrose (w/v), 0.8 mM boric acid, and 2 mM calcium nitrate – was added to a total volume of 2,500 µL. Each tube filled with PC or NC composition was mixed by brief vortexing, and pollen suspensions were then transferred into Petri dishes (30 mm in diameter). The dishes were incubated up to 2 h at 25°C in the dark. After incubation, 2 mL of pollen suspension was transferred to new 15 mL conical tube, and 1 mL of CR solution (5 mg·mL-1) was added. The samples were then incubated at room temperature (RT) for 30 min in the dark, and the optical density was measured at 600 nm using a UV/VIS spectrophotometer (JP/UV-2550, Shimadzu, Japan). The values obtained were corrected for background intensities by subtracting the spectra of either the PC or NC blank measured.
Flow Cytometry
Pollen at a concentration of 2.5 mg·mL-1 was suspended in liquid medium containing 10% sucrose (w/v), 0.4 mM boric acid, 1 mM calcium nitrate, and 2% DMSO (for PC) or 10% DMSO (for NC), and then incubated at 25°C for 4 h. Data were acquired by analyzing 1 mL of pollen culture per sample every hour using the Accuri C6 Flow Cytometer (Becton Dickinson, USA), according to the manufacturer’s instructions. Accuri C6 Flow Cytometer was kindly provided for this study by the Damyang Agricultural Technology Center. Events, represented by dots, were plotted along forward scatter (FSC) versus side scatter (SSC) parameters. The parameters FSC and SSC were proportional to the cell size and internal complexity, respectively. Pollen distributions were visualized by analyzing 50,000 events, acquired every hour, after culturing pollen in the growth media. The dot plot was divided into four quadrants (Q1-UL, Q1-UR, Q1-LL, and Q1-LR) to allow identification of the movement of dense population.
Results and Discussion
We first examined the germination rate of pear pollen in the solidified medium. As shown in Fig. 1, pollen tubes emerged after 1 h of incubation, and the tube length of the germinated pollens increased over the incubation periods, but except for the pollens that had already germinated, no further germination was observed. The germination rates in the solidified medium of 18.65, 23.52, and 21.37% were achieved after 1, 2, and 3 h of incubation, respectively. The germination rate may have been lower than expected because the viability of the pollen itself was low, but problems derived from the method of analyzing the germination rate cannot be ruled out.
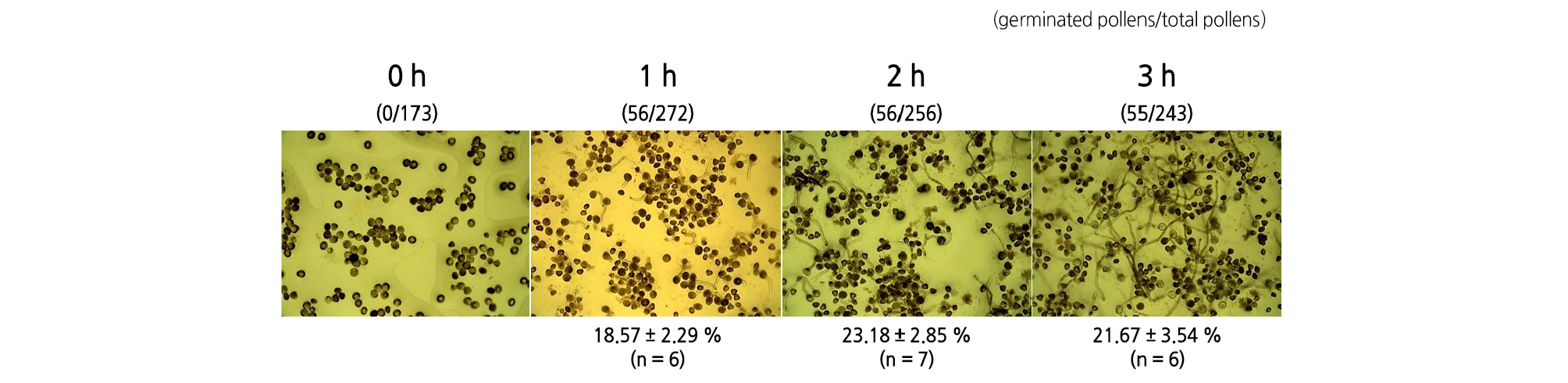
In vitro germination, which is used as a conventional method to test pollen vigor, involves counting the number of germinated pollens either directly under a microscope or after photographing the germinated pollen in order to examine the rate of pollen germination. This method is not only time-consuming and tedious, but also may be subjective because the counter is human, which may result in a lack of accuracy and reproducibility of the data obtained. In addition, when the pollen grains are either unevenly dispersed or aggregated with each other on the medium, the calculated germination rate may be lower than the actual germination rate. Therefore, accurately testing pollen germination through in vitro germination in solidified media has been shown to be difficult.
As a preliminary experiment for colorimetric assay, whether the degree of germination could be quantitatively measured by dividing pollen into positive control (PC) and negative control (NC) groups was confirmed by culturing pollen in liquid media. Whether there is a difference in germination rates between liquid and solidified media was also investigated. Germination of the PC group in the liquid medium was initiated after 1 h (27.01%), increased to 45.18% after 2 h, and reached 52.65% after 3 h (Fig. 2). Germination gradually increased with the incubation period up to hour three, and no further increase was observed during the rest of the incubation period. A difference in germination rate between the liquid medium and the solidified medium was observed: after 3 h of incubation, the germination rate in the liquid medium (52.65%) was 2.46 times higher than that in the solidified medium (21.37%).
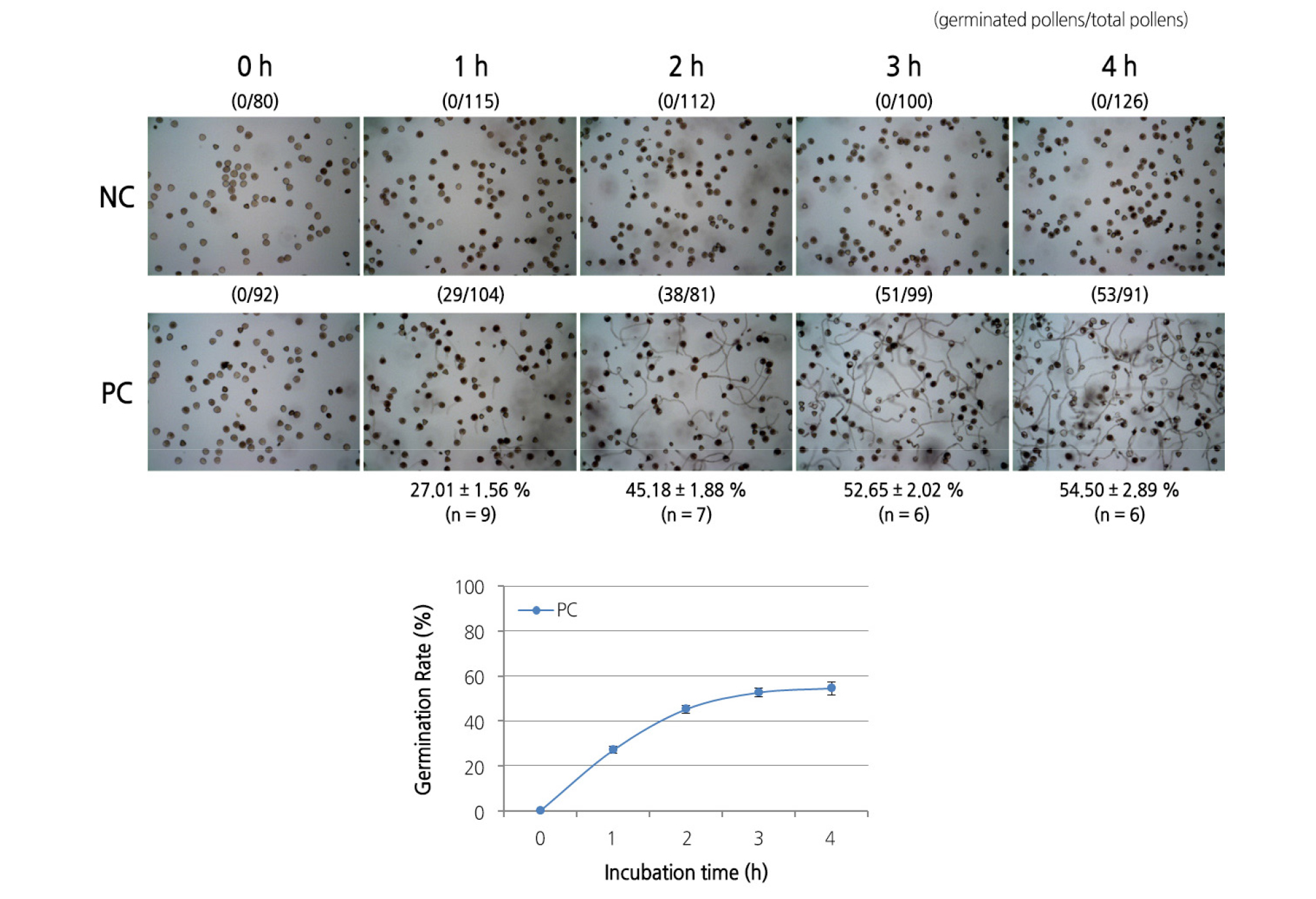
The difference in germination rate may be attributed to a problem that occurs during in vitro germination in solidified media. Pollen germination is considered to be affected not only by the viability of the pollen itself, but also by the density of pollen on the medium (Boavida and McCormick, 2007). In addition, the measured germination rate may be lower than the actual germination rate due to inaccurate measurements caused by the pollen tubes being entangled with each other. Therefore, the amount of pollen used to verify the pollen viability through in vitro germination should be adjusted to suit the observation and calculation of pollen germination.
When 10% DMSO (v/v) was added to the germination medium, the germination of pear pollen was completely inhibited, and no pollen tube was observed during incubation periods. These results are well described in a previous study of the inhibitory effects of DMSO on pollen tube initiation and tube growth. According to Dickinson and Cochran (1968), DMSO below 1% had little or no effect on the germination of lily pollen, but 5% DMSO prevented pollen tube initiation altogether. Although 5% DMSO is sufficient to prevent pollen tube initiation, the recovery of tube initiation and growth after the removal of DMSO suggests that DMSO reversibly inhibits the reactions required for tube initiation and growth. They concluded that the inhibition of pollen tube growth by DMSO is probably due to osmotic effects. The influence of DMSO on the osmotic effect was also explored through a quantitative analysis of the relative changes in cell volume through water permeabilization with DC-3F cells by de Ménorval et al. (2012). The quantitative analysis revealed a 9.4% (p < 0.001) reduction in the diameter of cells cultured in media containing 10% DMSO. The contraction of cells is the result of hyperosmotic shock due to increased osmotic pressure in media containing 10% DMSO.
To identify the previously observed pollen germination in liquid medium, a colorimetric assay was performed using Congo Red (CR) staining. Except for the absorbance from either pollen itself or non-germinated pollen, CR absorbance by elongated pollen tubes may be regarded as an indicator of germination. CR absorbance by elongated tubes may be calculated by subtracting the absorbance of the NC from that of the PC. CR absorbance of the PC or NC was measured in 1.25 to 10 mg·mL-1 of pollen (Fig. 3A). The absorbance of the PC increased as the concentration of pollen increased, while that of the NC did not change. This indicates that pollen tubes increased as the amount of pollen increased, thereby increasing the CR absorbance. The graph of the PC is expressed as a linear equation (y = 0.1574x+0.1425, R2 = 0.976) using the absorbance values of the pollen (precisely, the pollen tube) at 1.25 to 5.0 mg·mL-1 (Fig. 3C). When the concentration of pollen was 5.0 mg·mL-1 or more, the rate of increase in the width of the absorbance began to decrease; when the concentration was more than 7.5 mg·mL-1, the absorbance remained constant (Fig. 3B). Therefore, the concentration of pollen exceeding 5.0 mg·mL-1 was judged to be outside the meaningful range of the colorimetric assay, based on the absorbance value measured. Fig. 3B plots the difference in absorbance (Differ) between the PC and NC against various concentrations of pollen. The absorbance of Differ gradually increased at pollen concentrations of 1.25 to 5.0 mg·mL-1, and its trend line is expressed by the equation y = 0.127x + 0.0255, with R2 = 0.9938 (Fig. 3C). In the results of Fig. 3C, the slope of the equation can be interpreted as reflecting the actual germination rate of the pollen used in the assay, since the two variables of absorbance and the pollen concentrations are closely correlated (R2 = 0.9938), and the germination rate of the pollen used is theoretically the same within the effective range. Unfortunately, a formula for calculating the actual germination rate based on the slope of the equation obtained through the colorimetric assay has not yet been established. A further study using more pollen samples should be analyzed to obtain formulae that convert the slope value into an actual germination rate.
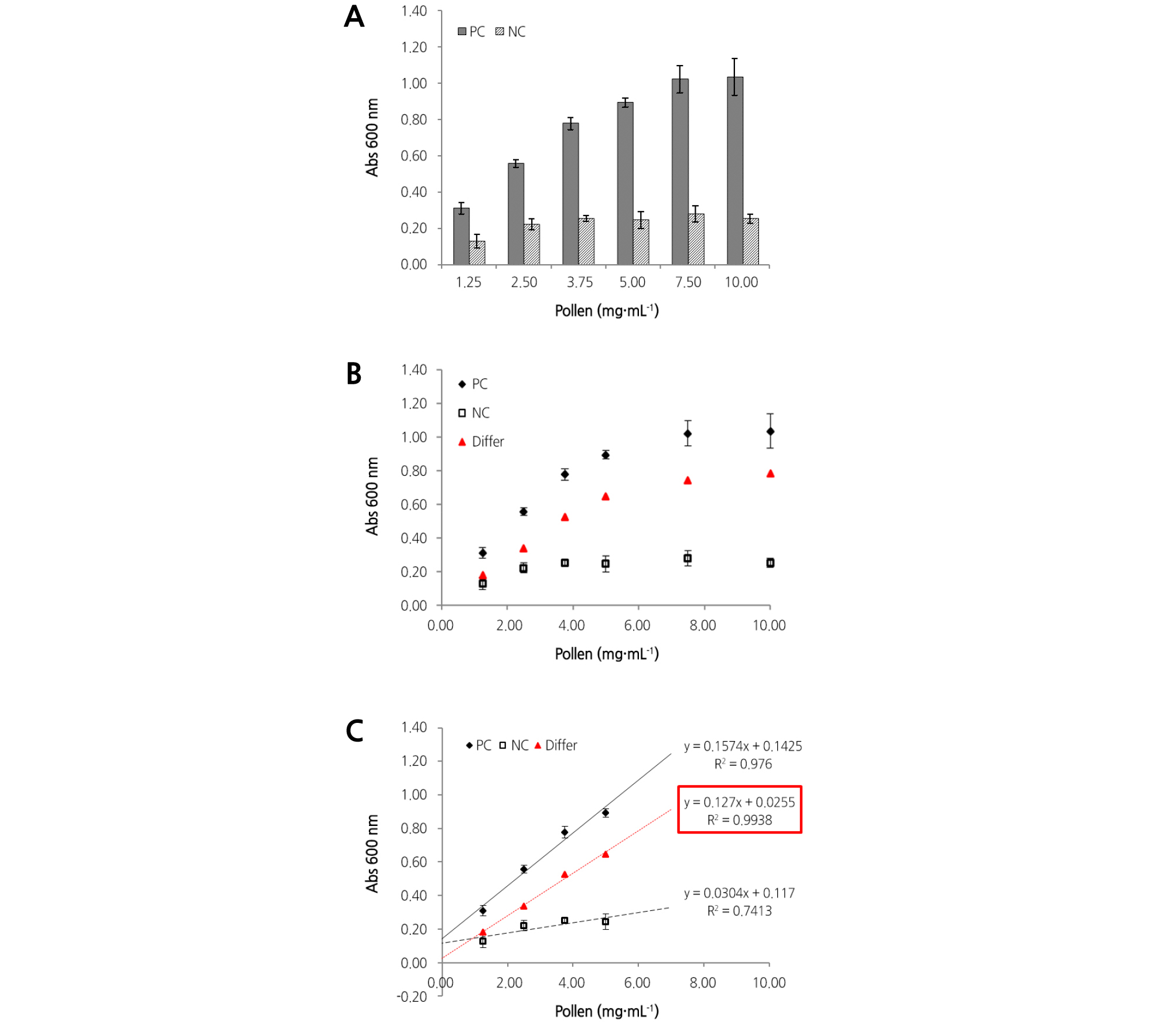
Fig. 3.
Congo Red (CR) absorbance at various concentrations of pollen in PC and NC. (A) Absorbance was plotted against various concentrations of pollen cultured in medium containing 2% DMSO (for PC) or 10% DMSO (for NC). Data represent the mean of CR absorbance with various concentrations of pollen examined, and vertical bars indicate the standard deviation of each mean, n = 6. (B) PC, closed diamonds; NC, open squares. Differ (closed triangles) represents the value calculated by subtracting the absorbance of the NC from that of the PC. (C) Trend lines for the PC (solid black line), the NC (dashed black line), and Differ (dotted red line). The equations of each trend line are listed on the right.
The cell wall of pollen tubes in most angiosperms is composed of cellulose (β-1,4-glucan), hemicellulose (mainly β- 1,4-xyloglucan), callose (β-1,3-glucan), and pectins (Mollet et al., 2013). In angiosperm pollen tubes, cellulose is generally present in low amounts, and instead callose is the main β-D-glucan found in the pollen tube cell wall (Qin et al., 2012). The content of callose was found to be as high as 81%, while that of cellulose was only 10% in Nicotiana pollen tubes grown for 4 h (Schlupmann et al., 1994).
In the colorimetric assay by Semedo et al. (2015), the specific interaction between CR and β-1,3-D-glucan exhibited a bathochromic shift of the maximum absorption from 488 to 516 (Semedo et al., 2015). Also, the fluorescence intensity of the dye plus β-D-glucan is much stronger than that of dye alone, and its absorption spectrum was not susceptible to reference chromophores (Wood and Fulcher, 1983). Fourier transform infrared (FTIR) analysis showed that CR is apparently bound to β-1,3-d-glucan by hydrogen bonds between hydroxyl groups of the polysaccharide chain and the amino groups of the dye, whereas sulphonic acid groups of dye do not participate this interaction (Semedo et al., 2015). Furthermore, except for β-1,3-d-glucan, the interaction between other polysaccharides (i.e. starch, cellulose, laminarin, and pectin) and CR did not exhibit any bathochromic shift of the maximum absorption (Semedo et al., 2015). These data suggest that the bathochromic shift is specific to β-1,3-d-glucan with a triple helix conformation, and the colorimetric assay using CR dye is suitable for quantitative measurement of newly elongated pollen tubes.
Quantitative methods for measuring pollen tube growth should be faster and more accurate than in vitro germination. Numerical expressions (such as equations) of pollen tube growth may be an important step towards establishing a quantitative method for verifying the quality of pollen. Additionally, after germination, the length of the elongated pollen tube is much higher than the diameter of the un-germinated pollen. The presence or absence of the pollen tube might be determined by flow cytometry, which can distinguish individual cells by their size or internal complexity. These concepts have motivated us to further investigate whether it is possible to verify pollen quality using flow cytometry.
We analyzed the pollen culture for the PC and NC groups every hour by flow cytometry, as shown in Fig. 4. Each dot on the graph represents one or more individual pollen grains on which a set of cytometric measurements were taken. The pollen distributions of the PC and NC groups before germination are almost similar, clustered in the Q1-LL quadrant (Fig. 4A). A difference in the pollen distributions between the PC and NC groups was observed 2 h after incubation. The ratio of the Q1-LR quadrant in the PC group increased in proportion to the incubation period, while that of the NC group remained almost unchanged. During pollen germination, the high-density region in the PC group on the dot plot forms a shifted transverse distribution to the right, as compared to that in the NC group (Fig. 4A). Fig. 4B shows the percentage changes in events of the PC and NC groups in the Q1-LR quadrant. The percentage changes in events of the PC group are plotted as a linear equation (y = 21.302x + 68.972) with R2 = 0.9009 over the incubation periods, while those of the NC show a low R-squared value (0.3264). This result suggests that the differences in the presence or absence of pollen tubes can be sufficiently discriminated by two parameters (i.e. FSC and SSC) of flow cytometry. The difference in the pattern of trend lines between the PC and NC is similar to that shown in Fig. 3C.
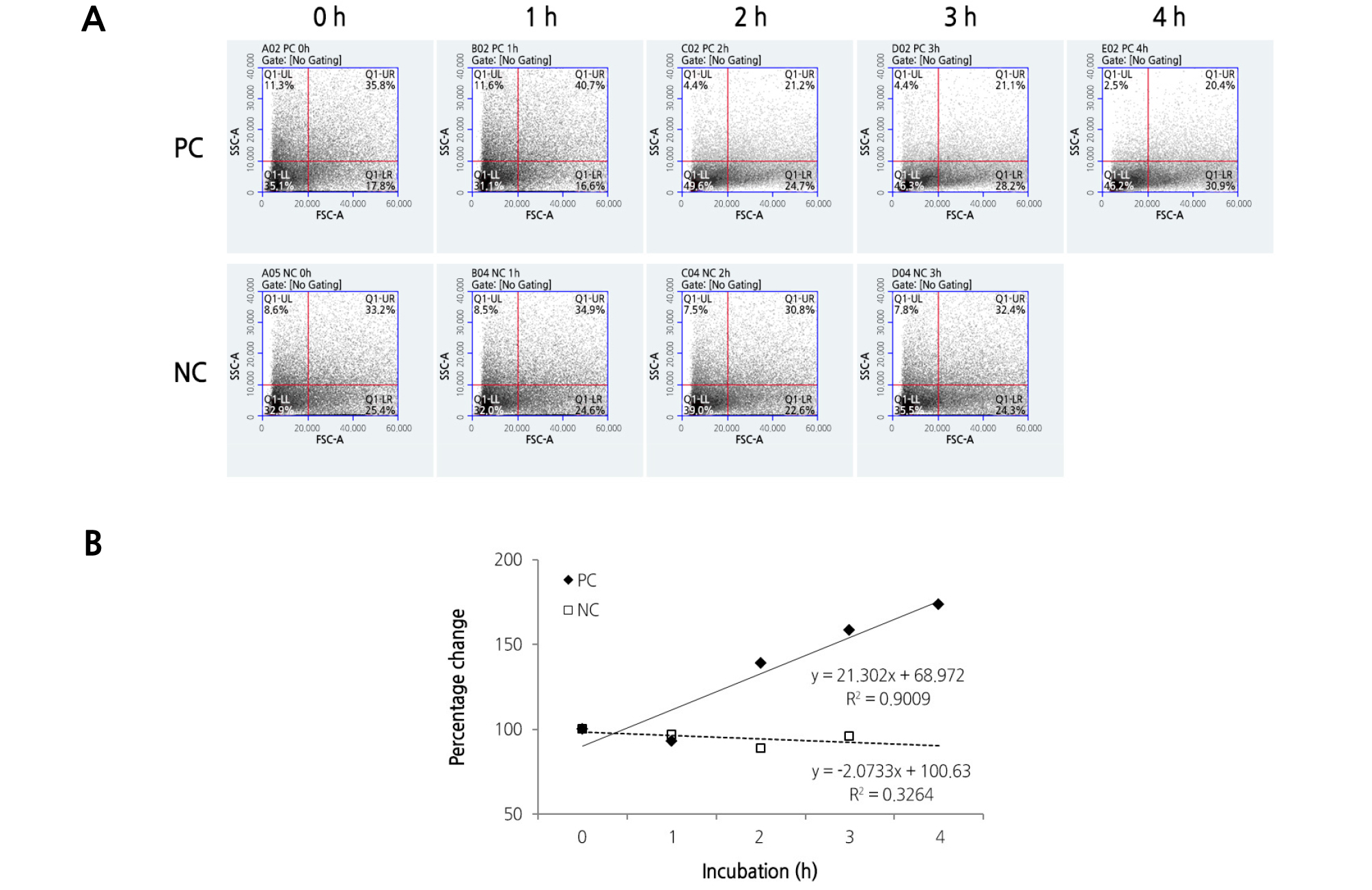
Fig. 4.
Identification of the change in pollen distribution during pollen germination by flow cytometry. (A) The upper and lower panels display the pollen distribution represented by dot plots under forward scatter (FSC) versus side scatter (SSC) parameters. The dot plots are divided into four quadrants (Q1-UL, Q1-UR, Q1-LL, and Q1-LR) to allow the movement of dense populations to be identified. Note the high-density region of the PC group on the dot plot forms a shifted transverse distribution to the right during pollen germination. (B) Closed diamonds and open squares indicate percentage changes in events of the PC and NC groups, respectively, in the Q1-LR quadrant. Trend lines for the PC (solid line) and NC (dashed line) groups were expressed, and their equations are listed on the right side.
Flow cytometry may be an alternative to manual microscope-based pollen counting, ensuring more reliable results in a fraction of the time needed for manual counting. Flow cytometers are used for a wide range of applications that include immunophenotyping, cancer diagnosis, ploidy analysis, cell sorting, and microbial identification (Lacombe and Belloc, 1996; Alvarez-Barrientos et al., 2000; Brown and Wittwer, 2000; Menten et al., 2009). Although flow cytometry can reduce user-dependent variance and improve the accuracy of measurement, inherent issues, such as clogging, are prone in fluidic-based systems (Schmid et al., 2003); the cost of the necessary equipment is also considerable. Despite its numerous advantages, flow cytometry is not yet widely used in horticultural sciences, and still limited to ploidy analysis (Kwon et al., 2017; Lee et al., 2017; Rhee et al., 2018), probably due to lack of access to the equipment and training required for this technique.
In the present study, we found that the differences in pollen distribution between the PC and NC groups in the Q1-LR quadrant and the percentage changes in PC group germination events could be expressed as a linear equation with a high R-squared value (0.9009). Based on these results, the degree of germination can be assumed by identifying the pollen distribution through cytometric analysis. However, 1 h after incubation, the percentage change in events in the PC group was out of the valid range of the equation, indicating a defect in this analysis that should be improved. Furthermore, correlation between the difference in pollen distribution and the actual germination rate still remains questionable, and our observations need to be confirmed using larger numbers of pollen samples.
In conclusion, this study shows that the germination rate of pear pollen differs depending on how the pollen is applied (i.e. scattered or suspended) to the medium. To establish a reliable method for verifying pollen germination, CR absorbance and patterns of pollen distribution were investigated. CR absorbance of “only” pollen tubes (Differ, as shown in Fig. 3C) against pollen within the effective range was expressed as a linear equation. Although the formula for calculating the actual germination rate has not yet been established, the slope of the equation obtained can be interpreted as reflecting the actual germination rate. Differences in pollen distribution between the PC and NC groups in the Q1-LR quadrant were also observed over the incubation periods.
These results demonstrate that monitoring the CR absorbance of newly elongated pollen tubes and the changes in pollen distribution along the FSC and SSC parameters will help verify pollen performance in pear plants. More evidence is needed to establish a formula that represents the actual germination rate of pollen, but our findings provide new insight into how to establish methods that ensure accurate and reliable validation of pollen germination.
